



Számtalan cikkünkben idéztük (lásd pl. EZT; összefoglalaló linkgyűjtemény ITT) már genetikusok tanulmányait a cigányok vérfertőzéséről, belterjességéről.
Lássuk a legújabb, ehhez kapcsolódó anyagot, amely az Ideggyógyászati Szemle 2007-es 1. számában jelent meg. Eredeti ITT. A tanulmány egyik legfontosabb megállapítása:
"A Lom-típusú neuropathiát csak roma közösségekben sikerült azonosítani Európa számos országában […] Négy magyarországi roma családnak összesen hat tagját vizsgáltuk […] Betegeink kétharmadában volt megfigyelhető enyhe mentális retardáció. "
Megismételjük: a vizsgált cigányok kétharmada szellemileg visszamaradott volt. Ráadásul ezt nem nácikok állítják valami nevenincs nácifasiszta honlapon, hanem genetikusok, ideggyógyászok az Ideggyógyászati Szemlében. Biztosan náci mind...
Van bárkinek bármi ellenvetése az ellen, hogy a deviáns cigányokat (azaz nem minden mokkát, “csak” egy részhalmazukat!) ennek megfelelően kezeljük például szavazási, választási jogosultság vagy a segélyek, ellátások kapcsán? Ahogy egy ötéves gyereknek se adunk százezres zsebpénzt (mert tudjuk, úgyis haszontalanságokra szórná el), így egy velük kb. azonos szellemi szinten álló (felnőtt) cigánynak sem érdemes pénzt adni. Ahogy a gyakorlatban bizonyíttatott, egyszerűen képtelen felelősen beosztani: rögtön elveri a kocsmában, bedobálja a játékgépbe, megveszi rajta a legdrágább csokit, kólát, chipset, aztán meg visít, hogy két nap után egy fillérje nem maradt.
Lássuk a legújabb, ehhez kapcsolódó anyagot, amely az Ideggyógyászati Szemle 2007-es 1. számában jelent meg. Eredeti ITT. A tanulmány egyik legfontosabb megállapítása:
"A Lom-típusú neuropathiát csak roma közösségekben sikerült azonosítani Európa számos országában […] Négy magyarországi roma családnak összesen hat tagját vizsgáltuk […] Betegeink kétharmadában volt megfigyelhető enyhe mentális retardáció. "
Megismételjük: a vizsgált cigányok kétharmada szellemileg visszamaradott volt. Ráadásul ezt nem nácikok állítják valami nevenincs nácifasiszta honlapon, hanem genetikusok, ideggyógyászok az Ideggyógyászati Szemlében. Biztosan náci mind...
Van bárkinek bármi ellenvetése az ellen, hogy a deviáns cigányokat (azaz nem minden mokkát, “csak” egy részhalmazukat!) ennek megfelelően kezeljük például szavazási, választási jogosultság vagy a segélyek, ellátások kapcsán? Ahogy egy ötéves gyereknek se adunk százezres zsebpénzt (mert tudjuk, úgyis haszontalanságokra szórná el), így egy velük kb. azonos szellemi szinten álló (felnőtt) cigánynak sem érdemes pénzt adni. Ahogy a gyakorlatban bizonyíttatott, egyszerűen képtelen felelősen beosztani: rögtön elveri a kocsmában, bedobálja a játékgépbe, megveszi rajta a legdrágább csokit, kólát, chipset, aztán meg visít, hogy két nap után egy fillérje nem maradt.
Kuruc.info - A.S.
ESETISMERTETÉSEK
Herediter motoros és szenzoros Lom-neuropathia
Első magyarországi közlés
Szabó Antal1, Siska Éva2, Molnár Mária Judit2
1Debreceni Egyetem, Orvos- és Egészségtudományi Centrum, Neurológiai Klinika, Debrecen
2Országos Pszichiátriai és Neurológiai Intézet, Molekuláris Neurológiai Osztály, Budapest
1Debreceni Egyetem, Orvos- és Egészségtudományi Centrum, Neurológiai Klinika, Debrecen
2Országos Pszichiátriai és Neurológiai Intézet, Molekuláris Neurológiai Osztály, Budapest
Levelező szerző (correspondent):
Dr. Molnár Mária Judit, Országos Pszichiátriai és Neurológiai Intézet, Molekuláris Neurológiai Osztály,
1021 Budapest, Hűvösvölgyi út 116. Telefon: (1) 391-5405, fax: (1) 391-5405. E-mail: molnarm@opni.hu
Dr. Molnár Mária Judit, Országos Pszichiátriai és Neurológiai Intézet, Molekuláris Neurológiai Osztály,
1021 Budapest, Hűvösvölgyi út 116. Telefon: (1) 391-5405, fax: (1) 391-5405. E-mail: molnarm@opni.hu
ÖSSZEFOGLALÁS
A herediter motoros és szenzoros neuropathia-Lom autoszomális recesszív, kizárólag az európai roma népességben előforduló, perifériás idegrendszert érintő betegség. Fő tünetei az első életévtizedben lassan progrediáló járászavar, amelyhez a második és harmadik évtizedekben a felső végtagi distalis izmok sorvadása, gyengesége, ízületi deformitások, később hypacusis is társul. A betegséget a 8q24 régióban elhelyezkedő NDRG1 gén homozigóta missense mutációja okozza. Egyes vizsgálatok eredményei szerint az NDRG1 gén mutációja a megváltozott lipidanyagcserén keresztül okozza a Schwann-sejtek működészavarát. Lom-neuropathia-fenotípust mutató romák esetében a genetikai vizsgálatok megelőzhetik a hagyományos invazív diagnosztikai módszereket, ezáltal gyorsítva és leegyszerűsítve a diagnózisalkotást, valamint a genetikai tanácsadást. Az örökletes neuromuscularis betegségek szűrése a közeljövőben fontos népegészségügyi feladattá válhat ebben a genetikailag még mindig izolált közösségben.
Kulcsszavak: herediter neuropathia, Lom, NDRG1
Érkezett: 2006. április 25.Elfogadva: 2006. szeptember 21.
A roma népcsoport közös eredettel és még mindig nagyfokú társadalmi izolációval jellemezhető populáció, ahol a szűkebb közösségen belüli házasság nem ritka jelenség. Populációgenetikai szempontból a cigányság földrajzilag szétszórt, de genetikailag izolált, viszonylag zárt közösségekben élő népcsoport. A nagyfokú genetikai izoláció miatt az autoszomális recesszív módon öröklődő betegségek gyakrabban fordulnak elő. Becslések szerint minden tizedik európai roma valamelyik örökletes neuromuscularis kórkép hordozója1. A többi európai népcsoporthoz képest az alábbi neuromuscularis kórképek fordulnak elő gyakrabban a romák körében: végtagövi típusú izomdystrophia 2C típusa (gamma-sarcoglycanopathia) mellett a congenitalis myasthenia szindróma 1A típusa, spinalis izom- atrophia, és háromféle herediter perifériás neuropathia, a herediter motoros és szenzoros neuropathia Lom és Russe formája, illetve a congenitalis cataracta facialis dysmorphia neuropathia szindróma (CCFDN)2.
Közleményünkben az 1990-es években, a Bulgáriában található Lom város mellett élő roma közösségben leírt herediter polyneuropathia klinikai és molekuláris genetikai jellemzőit mutatjuk be saját eseteink kapcsán. A Lom-típusú neuropathiát csak roma közösségekben sikerült azonosítani Európa számos országában is3, 4.
Módszerek
Négy magyarországi roma családnak összesen hat tagját vizsgáltuk herediter motoros és szenzoros neuropathia Lom-fenotípusa miatt. A betegek esetében részletes neurológiai vizsgálat történt. A perifériás idegek működését elektroneurográfiával követtük, a nervus ulnaris, nervus medianus, nervus peroneusok és nervus suralisok motoros és érző ágainak idegvezetését vizsgáltuk standard módszerekkel. A hat beteg közül három esetében koponya-MR-vizsgálat történt. Öt esetben agytörzsi akusztikuskiváltottválasz-vizsgálat (BAEP) készült az általánosan használt standard ingerléssel és regisztrálással. Két esetben nervussuralis-biopszia is történt, standard fénymikroszkópos és elektronmikroszkópos feldolgozással. A molekuláris genetikai vizsgálatokat mindegyik beteg esetében Perthben, az Edith Cowan Egyetemen, Ausztráliában végezték (Luba Kalaydjieva), az érintett génszakasz közvetlen szekvenálásával.
Eredmények
A klinikai tünetek mindegyik betegünk esetében már az első életévtizedben megjelentek. Az első markáns tünet a lassan progrediáló járászavar volt, a kézizmok gyengesége a második évtizedben vált nyilvánvalóvá. A vázizomtünetekkel párhuzamosan különböző ízületi deformitásokat is észleltünk, leggyakrabban a kéz kisízületeinek elváltozásai, az alsó végtagokon kalapácsujj, pes cavus és equinovarus volt megfigyelhető (1. ábra). Bár a neurológiai képet a motoros tünetek dominálták, betegeink zöménél mérsékelt fokban distalis típusú felszínes és mélyérzészavar is kimutatható volt. Két betegünk esetében hypacusis is megjelent, ezek a betegek már 20 évesnél idősebbek voltak. A perifériás idegek érintettsége mellett két betegünk esetében egyéb neurológiai tünetek is észlelhetők, mint dysphonia, egyoldali ptosis, fejtremor. Betegeink kétharmadában volt megfigyelhető enyhe mentális retardáció.
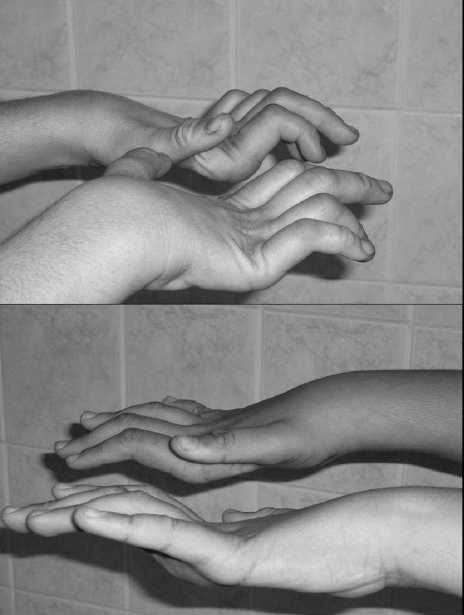 1. ÁBRA. HMSN-Lom-neuropathiás betegek esetében kifejezett a kéz kisízületeinek deformitása |
A műszeres diagnosztikai vizsgálatok közül az agytörzsi akusztikus kiváltott válasz vizsgálata öt betegből négy esetében mutatott működészavart. Elsősorban a nervus acusticus volt érintett, egy esetben a centrális hallópálya is károsodott. A koponya-MR-vizsgálat eredménye egy esetben volt kóros, enyhe kisagyi atrophiát jelzett.
Az elektroneurográfia mindegyik esetben súlyos demyelinisatiós motoros és szenzoros polyneuropathiát igazolt. Jellegzetes módon a szenzoros idegek ingerlésekor nem nyertünk válaszpotenciált, illetve a motoros idegekben súlyos vezetési zavart észleltünk.
Két esetben szövettani vizsgálat is történt. A nervus suralis félvékony metszeteiben a myelinhüvellyel rendelkező rostok kifejezett számbeli redukcióját láttuk. Aktív myelinhüvely-degenerációra és -regenerációra utaló jeleket nem figyeltünk meg, de egy esetben myelintörmeléket is sikerült kimutatni a Schwann-sejtek citoplazmájában.
A molekuláris genetikai vizsgálat során valamennyi esetben az NDRG1 génben (N-myc downstream-regulated gene 1) homozigóta R148X-pontmutációt lehetett igazolni az NDRG1 gén direkt szekvenálásával. A klinikai, elektrofiziológiai, morfológiai és genetikai vizsgálatok részletes eredményeit az 1. táblázatban mutatjuk be.
Megbeszélés
Saját betegeink adatai és az irodalomban eddig közölt esetek alapján a HMSN-Lom betegséget a következőkkel jellemezhetjük: A tünetek az első évtizedben jelentkeznek progrediáló járászavarral, az alsó végtagokon kialakuló distalis izomatrophia és vázizomgyengeség jeleként, majd ezt követi általában a második évtizedben a felső végtagi distalis vázizmok sorvadása. A motoros tünetek a kórlefolyás egésze alatt sokkal kifejezettebbek, mint a szenzoros tünetek. Az érzészavar az életminőséget érdemben nem befolyásolja. A tünetek korai kezdete is magyarázza, hogy csaknem mindig társul a kórképhez kéz- és lábízületi deformitás, karomállás, kalapácsujjak, pes cavus vagy pes equinovarus képében. Betegeink esetében ritkább deformitások is előfordultak, mint a scapula alata, illetve egy-egy esetben a perifériás neuropathiákban szokatlan neurológiai tünetként dysphonia, fejtremor is jelentkezett. A szenzoneurális hypacusis része a szindrómának, a harmadik életévtizedre szinte mindig kifejlődik a kisebb vagy nagyobb fokú hypacusis. A 20 évesnél idősebb betegeink közül háromból kettő esetében már kimutatható volt audiológiai vizsgálattal a halláscsökkenés. A hypacusis más örökletes idegbántalomnak is részjelensége lehet, ezért a kettő társulását nem lehet patognomikusnak tekinteni HMSN-Lom-ra5. Jellemző eltérések az I. csúcs latencia megnyúlása a nervus acusticus érintettsége jeleként, és a centrális interpeak latenciák megnyúlása agytörzsi károsodás jeleként. Betegeink felében enyhe mentális retardációra utaló jelek voltak megfigyelhetők, bár ez más leírásokban nem szerepel a kórkép részeként. A koponya-MR-vizsgálat általában normális, mi egy esetben enyhe cerebellaris atrophiát igazoltunk.
Az ENG-vizsgálat súlyos demyelinisatiós károsodásra utal. Jellemző, hogy a motoros vezetési sebességek súlyos fokban csökkennek, a motoros distalis latenciák jelentős egyidejű megnyúlásával. A szenzoros válaszok többnyire teljesen eltűnnek.
A nervus suralisból vett biopsziás minta szövettani vizsgálata során a myelinhüvellyel rendelkező rostok számának jelentős csökkenése figyelhető meg. Az aktív myelinhüvely-degeneráció és -regeneráció nem jellemző eltérések. Myelintörmelék előfordulhat a Schwann-sejtek citoplazmájában, korábbi demyelinisatióra utalva. Figyelemre méltó a myelinhüvely-károsodás mellett az axonok számának kifejezett redukciója is.
A betegség génjét 1996-ban azonosították6. A 8q24 régióban helyet foglaló NDRG1 gén homozigóta nonsense mutációja felelős a HMSN-Lom kialakulásáért7. Az R148X-pontmutáció founder mutációnak tekinthető, mivel gyakorlatilag ez az egy mutáció volt eddig kimutatható mindegyik HMSN-Lom-fenotípust megjelenítő beteg esetében. Az egyik nagyobb betegszámú vizsgálat során 104, a leggyakoribb mutációkra nézve negatív CMT-beteg közül két esetben találtak homozigóta NDRG1 R148X-mutációt és egy esetben egy másik pontmutációt, ami HMSN-Lom-fenotípushoz hasonló súlyosságú neuropathiával járt együtt. Az NDRG1 gén kódoló szekvenciájában nem találtak polimorfizmust, SNP (single nucleotide polymorphism) csak intronokban volt megfigyelhető8. Az NDRG1 gén terméke olyan intracelluláris protein, amely főleg stresszhatásra expresszálódik, például homocisztein- vagy 2-mercaptoetanol-, illetve nehézfém-expozíció után, de megfigyelték, hogy p53-expresszió vagy DNS-károsodás is indukál NDRG1-expressziót9. A sejtek növekedési fázisában gátolt az NDRG1-expresszió. Kísérletes adatok alapján úgy tűnik, hogy az NDRG1 gén fontos szerepet játszik a sejtszinten végbemenő stresszválasz-mechanizmusokban, illetve a sejtdifferenciálódásban, mind normális, mind patológiás körülmények között, például daganatsejtekben10.
Ami a központi és perifériás idegrendszert érinti, az NDRG1 gén kifejezetten expresszálódik a Schwann-sejtek citoplazmájában, kisebb mértékben az oligodendrogliában, a motoros és szenzoros neuronokban viszont nem expresszálódik. Ez összhangban van a hisztológiai eltérésekkel, amely elsődleges demyelinisatióra utal11.
NDRG1-re génkiütött egérben progresszív demyelinisatiós neuropathia fejlődött ki12. Nem ismert a pontos molekuláris mechanizmus, amely a HMSN-Lom-fenotípushoz vezet, de egyes adatok szerint a Schwann-sejtek lipidanyagcseréjében alakul ki működészavar, az APOA-1 és APOA-2 szállító fehérjék megváltozott működése miatt.
Az APO A-1 és APO A-2 a HDL (high density lipoprotein) legfontosabb alkotórészei közé tartoznak. Kísérletes bizonyítékok vannak az NDRG1, az APO A1 és APO A2 közötti interakcióra. Bár a folyamat minden lépése még nem ismert, a fenti fehérjék közötti kapcsolat a koleszterin-homeosztázisban és a sejten belüli lipid- és fehérjetranszportban játszik feltehetőleg szerepet. Erre utal indirekt módon az is, hogy az NDRG1 R148X genotípust hordozók közül a férfiakban szignifikánsan alacsonyabb a HDL-koleszterin-szint13. Az eltérés nem specifikus jellegének hátterében valószínűleg az eltérő hormonális hatások állnak. Az általunk vizsgált hat beteg közül kettő fiú, mindkettőjük HDL-koleszterin-szintje jóval a normáltartomány alatt volt (0,69 mmol/l és 0,79 mmol/l, normálisnak mondható az 1 mmol/l feletti HDL-koleszterin-szint), utalva a megváltozott lipidanyagcserére. Ez további kutatásokat ösztönözhet később az NDRG1 atherosclerosisban betöltött lehetséges szerepére vonatkozóan.
Az autoszomális recesszív módon öröklődő herediter neuropathiák genetikai diagnosztikája az utóbbi években jelentősen gazdagodott. Az NDRG1 mutációja mellett többek között a myotubularin-related protein-2 (MTMR2), GDAP1 (ganliozid indukálta differenciációasszociált protein), KIAA1985 és periaxin gének mutációi okoznak demyelinisatiós fenotípussal járó autoszomális recesszív herediter neuropathiát14, 15. Világszerte ilyen irányú kiterjedt genetikai vizsgálatokat csak kevés, elsősorban nem rutindiagnosztikával foglalkozó kutatólaboratóriumokban végeznek, ezért a mindennapos klinikai gyakorlatban gyakran nem sikerül pontos genetikai diagnózishoz jutni. A pontos klinikai tünetek és az idegbiopsziával nyert szövettani eredmény birtokában lehet tovább szűkíteni a vizsgálandó gének körét, ami esélyt ad a végleges molekuláris diagnózisra, és talán a jövőben a pontos genetikai diagnózis a genetikai tanácsadásban betöltött szerepe mellett a terápia megválasztásában is segíthet majd.
Jelen esetben a betegek etnikai hovatartozása is segíthet a betegség etiológiájának tisztázásában. Roma populációban érdemes a leírt fenotípus esetén a HMSN-Lom-neuropathiára gondolni. Az említett sajátosságok miatt a genetikai vizsgálat megelőzheti, esetleg fel is válthatja a jelenleg alkalmazott invazívabb diagnosztikai módszereket. Ilyen példa már létezik, a neuromuscularis betegségek közül a facioscapulohumeralis izomdystrophia esetében a molekuláris genetikai vizsgálat már régen felváltotta az izombiopsziát és EMG-vizsgálatot.
Mindezek alapján a neuromuscularis betegségek szűrése és az ezzel kapcsolatos genetikai tanácsadás fontos közegészségügyi feladattá válhat a jövőben, mert a genetikailag még mindig zárt, nemritkán szűkebb közösségen belül házasodó populációban a jövőben is az autoszomális recesszív módon öröklődő kórképek gyakori előfordulása várható.
KÖSZÖNETNYILVÁNÍTÁS
A molekuláris genetikai vizsgálatok elvégzéséért köszönetet mondunk Luba Kalaydjievának, aki a vizsgálatok időpontjában az ausztráliai Edith Cowan Egyetem munkatársa volt. A vizsgálatokat az ETT 179/2003 pályázat támogatta.
Irodalom
- Kalaydjieva L, Lochmüller H, Tournev I, Baas F, Beres J, Colomer J, et al. Workshop report 125th International Workshop: Neuromuscular Disorders in the Roma (Gypsy) Population, 23-25 April 2004, Naarden, The Netherlands. Neuromuscular Disorders 2005;15:65-71.
- Navarro C, Teijeira S. Neuromuscular disorders in the Gypsy ethnic group. A short review. Acta Myol 2003; 22(1):11-4.
- Kalaydjieva L, King R, Gresham D, Molnar M, Tournev I, Angelicheva D, et al. Hereditary motor and sensory neuropathy Lom. Acta Myologica 2001;20:192-201.
- Kalaydjieva L, Nikolova A, Turnev I, Petrova J, Hristova A, Ishpekova B, et al. Hereditary motor and sensory neuropathy-Lom, a novel demyelinating neuropathy associated with deafness in gypsies clinical, electrophysiological and nerve biopsy findings. Brain 1998;121:399-408.
- Raglan E, Prasher DK, Trinder E, Rudge P. Auditory function in hereditary motor and sensory neuropathy (Charcot-Marie-Tooth disease). Acta Otolaryngol 1987;103(1-2): 50-5.
- Kalaydjieva L, Hallmayer J, Chandler D, Savov A, Nikolova A, Angelicheva D, et al. Gene mapping in gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nat Genet 1996;14:214-7.
- Kalaydjieva L, Gresham D, Gooding R, Heather L, Baas F, de Jonge R, et al. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet 2000;67(1):47-58.
- Hunter M, Bernard R, Freitas E, Boyer A, Morar B, Martins IJ, et al. Mutation screening of the N-myc downstream-regulated gene 1 (NDRG1) in patients with Charcot-Marie-Tooth Disease. Hum Mutat 2003;22(2):129-35.
- Kurdistani SK, Arizti P, Reimer M, Sugrue S, Aaronson A, Lee SW. Inhibition of tumour cell growth by RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res 1998;58:4439-44.
- van Belzen N, Dinjens WN, Diesveld MP, Groen NA, van der Made AC, Nozawa Y, et al. A novel gene which is up-regulated during colon epithelial cell differentiation and down-regulated in colorectal neoplasms. Lab Investig 1997;77:85-92.
- Berger P, Sirkowski E, Scherer S, Suter U. Expression analyis of the N-myc downstream-regulated gene 1 indicates that myelinating Schwann cells are the primary disease target in hereditary motor and sensory neuropathy-Lom. Neurobiology of Disease 2004;17:290-99.
- Okuda T, Higashi Y, Kokame K, Tanaka C, Kondoh H, Miyata T. NDRG1-deficient mice exhibit a progressive demyelinating disorder of peripheral nerves. Mol Cell Biol 2004;24(9):3949-56.
- Hunter M, Angelicheva D, Tournev I, Ingley E, Chan D, Watts G, et al. NDRG1 interacts with APO A-I and A-II and is a functional candidate for the HDL-C QTL on 8q24. Biochem Biophys Res Commun 2005;332(4):982-92.
- Vallat JM, Grid D, Magdelaine C, Sturtz F, Tazir M. Autosomal recessive forms of Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep 2004;4(5):413-9.
- Parman Y, Battaloglu E, Baris I, Bilir B, Poyraz M, Bissar-Tadmouri N, et al. Clinicopathological and genetic study of early-onset demyelinating neuropathy. Brain 2004;127 (Pt11):2540-50.
HEREDITARY MOTOR AND SENSORY NEUROPATHY-LOM – FIRST HUNGARIAN CASE REPORT
Szabó A, MD; Siska É, MD; Molnár MJ, MD, PhD
Ideggyogy Sz 2007;60(1–2):51–55.
Szabó A, MD; Siska É, MD; Molnár MJ, MD, PhD
Ideggyogy Sz 2007;60(1–2):51–55.
Hereditary motor and sensory neuropathy-Lom is an autosomal recessive disorder of the peripheral nervous system, which occurs only in the european Roma population. The symptoms start in the first decade with slowly progressive gait disturbance, weakness and wasting of distal upper extremity muscles, joint deformities and hearing loss develop later in the second and third decades. This disorder is caused by a homozygous missense mutation of the NDRG1 gene, located in the 8q24 region. The Schwann cell dysfunction is most probably caused by altered lipid metabolism as a consequence of the NDRG1 mutation. Molecular genetic testing can be a first diagnostic step among roma individuals showing a Lom neuropathy phenotype, making evaluation of such patients and also genetic counselling faster and easier. Screening for hereditary neuromuscular disorders in this genetically isolated community may become an important public health issue in the near future.
Keywords: hereditary neuropathy, Lom, NDRG1




